微粒子計數器相關
安全的數據完整性等於輕鬆合規

TSI 粒子計數器能夠在專為GMP 使用者設計的數據完整性(DI) 模式下運行——符合 ALCOA+ 原則的完整、一致和準確的數據。
通過顆粒計數數據實現這一目標需要使用專為符合 GMP 標準而設計的顆粒計數器,並結合良好的測試程序。
TSI 在選定型號的粒子計數器上,提供增強的數據完整性功能,這些儀器與 TSI TrakPro Lite Secure (TPLS) 軟體配合使用,提供一種輕鬆實現數據完整性合規性的途徑。
TPLS 軟件專為符合FDA 21 CFR Part 11標準而設計,所有型號 AeroTrak 監控粒子計數器、微粒子計數器和 BioTrak 即時微生物粒子計數器的標準配置,可滿足 FDA 21 CFR Part 11 法規要求。
其他微粒子計數器試圖通過在儀器上添加第 11 部分功能來解決數據完整性問題,然而,TSI 數據完整性粒子計數器通過使用 TPLS 軟件作為用戶管理、儀器配置和報告生成的單一集中位置,提供了優於這些第 11 部分粒子計數器的優勢,這消除了必須在每個單獨的儀器上執行這些任務的麻煩,就像儀器上具有第 11 部分功能的其他粒子計數器一樣。
TSI 微粒子計數器全系列產品拓生科技總代理
法規類相關小知識
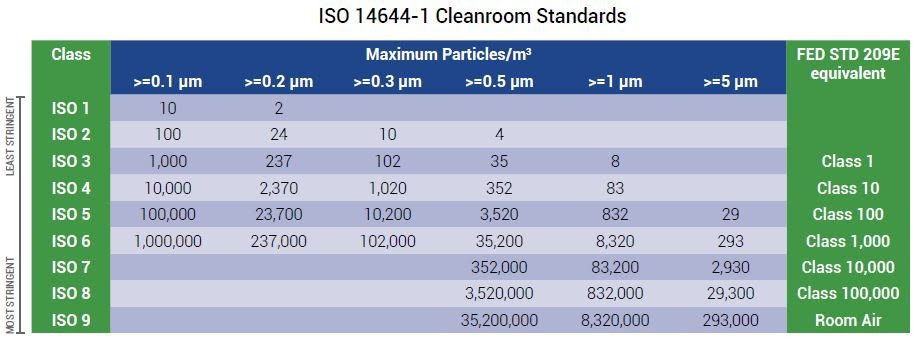
什麼是 ISO 8 無塵室分類?
每個潔淨室級別都用每立方米或立方英尺空氣中的最大顆粒濃度表示,ISO 8 是第二低的潔淨室等級
ISO 14644-1 分類潔淨室是一個房間或密閉環境,在其中保持低顆粒數至關重要,通常,這些顆粒是灰塵、空氣中的微生物、氣溶膠顆粒和化學蒸汽,除了粒子計數之外,潔淨室通常還可以控制許多其他參數,例如壓力、溫度和濕度,此外,要被視為潔淨室,該空間需要高效微粒空氣 (HEPA) 過濾器來去除空氣中的顆粒。
ISO 14644-1 潔淨室分為 ISO 1 到 ISO 9,每個潔淨室級別都用每立方米或立方英尺空氣中的最大顆粒濃度表示,ISO 8 是第二低的潔淨室等級。
ISO 8 潔淨室標準
ISO 8 潔淨室允許的顆粒數取決於參考標準及其測量值,根據美國聯邦標準 209E,ISO 8 級潔淨室也稱為 100,000 級潔淨室。
2017-08-10 ISO潔淨室標準
ISO 14644-1 取代了聯邦標準,並聲明 ISO 8 潔淨室每立方米空氣中需要少於 3,520,000 個 ≥ 0.5 微米大小的顆粒,在 ISO 8 潔淨室中只能測量 0.5 微米或更大的顆粒,這樣做是因為較小粒徑的濃度太高。
ISO 8 保護要求和設計注意事項
設計潔淨室需要根據行業和應用考慮額外的監管標準和要求,但是,對於 ISO 8 潔淨室,需要考慮幾個一般要求和環境參數,對於 ISO 8 潔淨室,這些包括:
- HEPA過濾
- 每小時換氣次數 (ACH)
- 空氣壓力
- 溫度和濕度
- 在該空間工作的人員數量
- 靜電控制
- 燈光
- 噪音水平
由於 ISO 8 潔淨室專注於測量 0.5 微米或更大的顆粒,因此 HEPA 過濾系統的效率需要達到 99.97%,建議每小時至少換氣 20 次,空氣的最終過濾發生在空氣進入潔淨室的地方,此外,ISO 8 潔淨室最常見的是採用非單向或混合氣流模式,牆壁空氣回流率較低。
關於 ISO 8 的常見問題解答
ISO 7 和 ISO 8 有什麼區別?
ISO 7 和 ISO 8 潔淨室之間的兩個主要區別是顆粒計數和 ACH 要求,這使它們在不同的應用中脫穎而出。
ISO 7 潔淨室必須有 352,000 個≥0.5 微米/立方米的顆粒和每小時 60 個 ACH
ISO 8 為 3,520,000 個顆粒和 20 個 ACH。
什麼時候需要潔淨室?
潔淨室可用於許多行業和應用,對於清潔和無菌至關重要的空間,它們是必需的,就上下文而言,ISO 8 潔淨室通常比典型的辦公環境清潔 5-10 倍,具體來說,在醫療器械和藥品製造潔淨室中,產品的安全和質量至關重要,如果進入空間的顆粒過多,原材料、製造過程和成品都會受到影響。
環境監測系統旨在收集、分析和通知詳細的潔淨室環境數據,特別是對於製造空間,潔淨室監控的目標是評估產品的潛在污染風險並保持符合監管標準。
什麼是 ISO 8 潔淨室分類?
FMS 是 TSI的連續環境監測軟體,可從室內潔淨室測量傳感器收集數據,可以監控多個位置的不同設施,提供即時環境數據訪問和復雜的分析,能在軟件中直觀地看到實時警報通知,並通過短信和電子郵件發送給用戶,即時、最新的報告和圖表,TSI 的 FMS 有助於在生產製造過程中持續監控環境,以簡化操作、最大限度地降低污染風險並提高產品質量。
- 醫療器械製造
- 藥物製造和合成
- 半導體製造
- 電子製造
FMS 幫助潔淨室滿足 ISO 14644、USP<797>、USP<800>、cGMP、歐盟附件 1、GAMP、聯合委員會、FDA 21 CFR 第 11 部分、歐盟附件 11 和 GAMP5 的必要監管要求。
全廠遠距即時監測軟體(FACILITY MONITORING SYSTEM, FMS)<----------請點我
GMP附件(1) 2022 更新細目:第 1 部分

歐盟 (EU) 人用和獸用醫藥產品良好生產規範 (GMP) – 附件 1,通常稱為 GMP 附件 1,最初於 2017 年作為草案發布。該草案讓許多從事製藥行業的人業界對一些似乎缺乏細節和完整性的法規擬議修改感到疑惑。
現在,GMP Annex 1 已經發布,並附有解釋,此次更新預計會改變歐盟的指導方針,還會改變美國 FDA 的規定,我們應該期待在全世界看到此更新後的影響。
此新法規更新於 2022 年 8 月 25 日發布。
附件 1 中的主要變化圍繞著通過詳細和廣泛的污染控制策略 (CCS) 進行質量風險管理 (QRM) 的基礎設施方法。
我們在對清潔區之間的移動、人員存在和文件要求的嚴格限制中看到了這一點。一般來說,這將需要對使用無法跟上新指南的舊設備的操作進行廣泛且的調整。
此篇中,我們分解附件 1 的 10 個部分前 5 個部分,對每個部分進行簡要概述,以提供對新法規的基本了解,第 2 部分中,我們將介紹剩下5個部分。
GMP 附件 1 第 1 部分:
第 1 部分定義了文檔的範圍和意圖。附件 1 旨在解決“應用質量風險管理 (QRM) 原則生產所有無菌產品的設施、設備、系統和程序的設計和控制”。作者確實注意到,雖然這是為了為無菌產品製定標準而編寫的,但有些部分只是適用於任何潔淨室的良好做法。
GMP 附件 1 第 2 部分:
第 2 部分標題為“原則”,討論了附件 1 背後的思想基礎。本質上:無菌產品應在專為無菌設計的環境中由訓練有素的員工使用有意識的、可追溯的過程生產。
雖然理論上很簡單,但這部分是我們開始看到附件 1 的新主題的地方:自動化以盡可能避免污染,包括從生產線上撤離人員。
人類是潔淨室的主要污染源——這不足為奇。因此,更多依賴機器和自動化是有道理的,但隨著這些技術的進步,圍繞文檔、驗證、安全、數據完整性和可追溯性的法規和指南也越來越多。
GMP 附件 1 第 3 部分:
第 3 部分深入探討製藥質量體系 (PQS),它概述了良好 PQS 的基礎:
- 它應該集成到產品生命週期的所有階段。
- 製造商應該以這樣的方式了解產品,以便他們能夠識別出什麼時候出了問題並使無菌處於危險之中。
- 如果設備出現故障,應進行適當的調查,最後採取糾正和預防措施 (CAPA)。
- 嚴格應用和遵循風險管理,以避免所有類型的污染。
- 高級管理層應參與監控和定期審查風險管理策略。
- 完成、儲存和運輸產品不應使其處於危險之中。
- 負責驗證和分銷產品的人員應有權訪問生產和質量信息,他們還應該以這樣的方式了解流程,以便識別何時出現可能影響最終產品質量的問題。
GMP 附件 1 第 4 部分:
第 4 部分概述了對潔淨室設計的期望,包括限制進入屏障系統 (RABS)、氣閘、過濾器和更衣室的使用,此章節還深入探討了無塵室應如何設計,例如使用的材料、天花板形狀等,隨著 RAB 和隔離器等附加技術的引入,以協助污染控制策略 (CCS),我們看到另一個附件 1 的主要變化之一。
在第 4 部分中,我們看到介紹了 4 個等級的潔淨區:A、B、C 和 D 級。
A 級被認為是房間中最潔淨的部分,用於高度敏感的過程,例如無菌灌裝。
B 級區域用作 A 級區域的背景室:本質上,與 C 級和 D 級進一步分離。
C 級和 D 級區域用於製造過程中不太關鍵的階段。
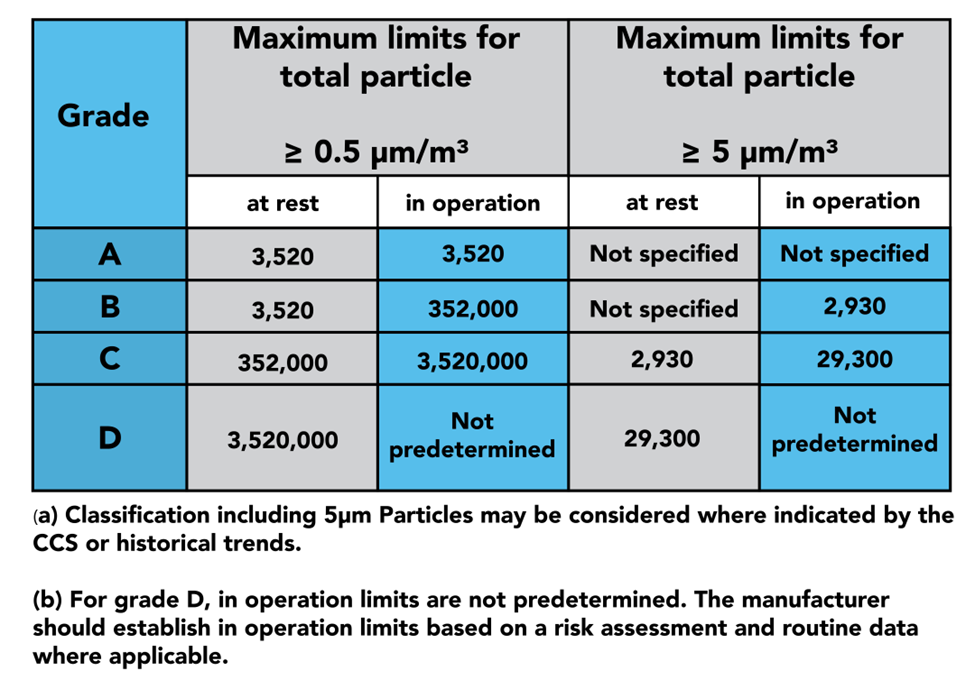
在設計潔淨室時,應該考慮物體、人員和空氣如何從一個區域傳遞到另一個區域,因為這是主要的污染源。
附件 1 第 4 部分概述瞭如何以最小化這種風險的方式執行這些過程,這是通過使用 RABS、隔離器、氣閘、過濾器、正壓室和負壓室等來完成的。
第 4 部分的後半部分深入探討了區域的資格和驗證以及用於監視它們的設備,如上表所示,GMP 規定應在靜止狀態和運行狀態下進行鑑定。在這裡您可以找到有關樣本大小和位置的說明,以正確認證您的潔淨室。
GMP 附件 1 第 5 部分:
第 5 部分讓您了解如何恰當地描述和記錄您的設備和流程。它還解決了對合格粒子計數器的需求。
這部分還規定了所用設備的重要性和正確清潔設備,以及如何在區域之間使用設備,例如,傳送帶不應在區域之間通過,因為這會帶來污染風險。
EUGMP Annex1 2022 <----------- 請點我
GMP 附件 1 2022 更新細目:第 2 部分
通過全面的污染控制策略 (CCS),更加關注質量風險管理 (QRM) 是本文件中最大的變化和轉變,一如既往,GMP 的目標是提供安全的產品並保持人們的健康,技術進步減少了製造或灌裝線上對人工干預和人員的需求,最大限度地減少或消除人員可以大大降低污染風險,但確實需要更改 SOP 和文檔,隨著技術的發展,必須調整您維護的方式,以確保所有信息和數據的完整性。
GMP 附件 1 由 10 個部分組成,涉及藥品的無菌工藝,儘管本文件的許多部分可作為其他行業的優秀指南。此是我們系列文章的第二部分,它分解了 10 個部分中每一部分的基礎知識,並強調了理論和實踐方面的一些顯著變化。第 1 部分涵蓋前 5 部分,此篇涵蓋後半部分:第 6 部分到第 10 部分。
GMP 附件 1 第 6 部分:
GMP 附件 1 的第 6 部分討論了無塵室中的公用設施,特別是那些與最終產品接觸並構成污染風險的公用設施,例如,水和蒸汽,它還列出了應該在無塵室中的整個生命週期記錄的實用程序。
該文件的這一部分專門針對 4 個公用設施:水系統、用於消毒的蒸汽、氣體和真空系統,以及加熱、冷卻和液壓系統。
水可能是最密集的公用事業,因為註射用水 (WFI) 受到嚴格的標準約束,這確實討論了 WFI 的生產和儲存,以及水處理廠的重要性和設計。
蒸汽應來自與水保持相同標準的水源。它不應包含可能對產品造成傷害的添加劑。
與產品接觸的氣體應經過適當過濾和監測。應記錄並考慮所有相關參數,例如油接觸。
最後,加熱、冷卻和液壓系統應盡可能位於潔淨室外面。
GMP 附件 1 第 7 部分:
第 7 部分概述了有關潔淨室人員的規定,包括潔淨室著裝和行為方面的培訓,雖然附件沒有定義培訓期間之間的時間,但它確實規定培訓應該“定期”進行,並強調理解的重要性。
本節繼續概述他們的健康發生變化時的溝通、良好的衛生習慣,以及根據他們所在的潔淨室級別在無菌長袍下穿(或不穿)的衣服類型在工作。
為了與減少潔淨室中對人類的依賴這一主題保持一致,我們還被指示在任何給定的潔淨區內應盡可能減少人員數量,此外,在潔淨室的設計過程中應記錄允許的最大數量。
附件 1 的這一部分顯示了與先前版本相比的其他更改。對潔淨室人員知識的關注度更高,這也反映在第 3 部分中。
GMP 附件 1 第 8 部分:
第 8 節是附件 1 中最全面的部分,因為它實際上概述了無菌產品的生產和相關技術。這部分專門針對:
- 最終滅菌產品
- 無菌製備和加工
- 無菌產品的整理
- 消毒
- 加熱殺菌
- 濕熱滅菌
- 乾熱滅菌
- 輻射滅菌
- 環氧乙烷滅菌
- 無法在最終容器中滅菌的產品的過濾滅菌
- 成型-填充-密封 (FFS)
- 吹-灌-封
- 冷凍乾燥
- 封閉系統
- 一次性系統 (SUS)
對於這些方面中的每一個方面,我們都看到了對將 CCS 貫穿始終的高度重視。根據您使用的工藝和技術,我們發現使用前滅菌後完整性測試 (PUPSIT)、特定處理保持時間、目視檢查、關鍵干預後加強的無菌測試抽樣以及容器密閉完整性的要求發生了變化。
GMP 附件 1 第 9 部分:
第 9 部分可能是 Lighthouse Worldwide Solutions 中我們最喜歡的部分之一:環境和過程監控。從一開始,我們就看到您更加關注將 CCS 納入您的環境監測中,這通常包括總粒子監測;活粒子監測;溫度、濕度等特定特性監測;無菌過程模擬 (APS) 監控。
附件 1 的這一部分準確地概述了您應該如何監控您的潔淨室 – 並記錄所有列出的因素,對於不同等級的潔淨室(A、B、C 和 D 級,A 級是最乾淨的),應制定不同的監控協議,例如,需要連續監測 A 級潔淨區的總顆粒物。
我們還會獲得一份詳細的項目列表,其中列出了在您的 APS 設計中需要考慮的事項,雖然 APS 本身不應該驗證潔淨室,但它是一項定期進行的關鍵檢查,應該這樣對待。
GMP 附件 1 第 10 部分:
最後,附件 1 涵蓋了質量控制 (QC),我們在其中看到了本文檔所做的轉變,這包括關注圍繞 CCS 的綜合人員知識和發展,總體而言,附件 1 的新草案增加了測試負擔並減少了應實際參與無菌過程的人員數量。
EUGMP Annex1 2022 <----------- 請點我
歐盟 GMP 附件1和 ISO 14644-1 的協調:深入了解
來自 Invar 的 Alexander Fedotov 解釋說,標準的協調似乎是設定顆粒限制的驅動力,以及為什麼這可能並不總是導致合乎邏輯的科學決策
歐盟 GMP 附件 1 規定了粒徑 > 0.5 µm 和 > 5.0 µm 的空氣傳播顆粒濃度限值,美國 FDA 僅要求對 0.5 µm 的那些進行控制,沒有證據表明美國的藥物更糟糕,控制 > 5.0 µm 的顆粒是否合理,或者這是一個沒有被現代知識證實的教條?
除了 > 0.5 µm 之外,是否值得控制 > 5.0 µm 的顆粒?此篇將討論錯誤解釋 ISO 14644-1 的後果並解釋其起源。
對歐盟 GMP 附件 1 的修改
FDA 關於無菌空氣潔淨度的規範自 1980 年代以來一直很穩定,但歐盟 GMP 附件 1 已多次更改(表 1),如表 1(A/B 級)中標記的單元格所示,> 5.0 µm 濃度的顆粒限值可能會發生變化,直到 2003 年,初始限制為零,因為這些區域不允許存在微生物,有專家說0在統計上是無意義的,單次計數可能是電子噪聲的結果,這意味著2003年進行了更改。
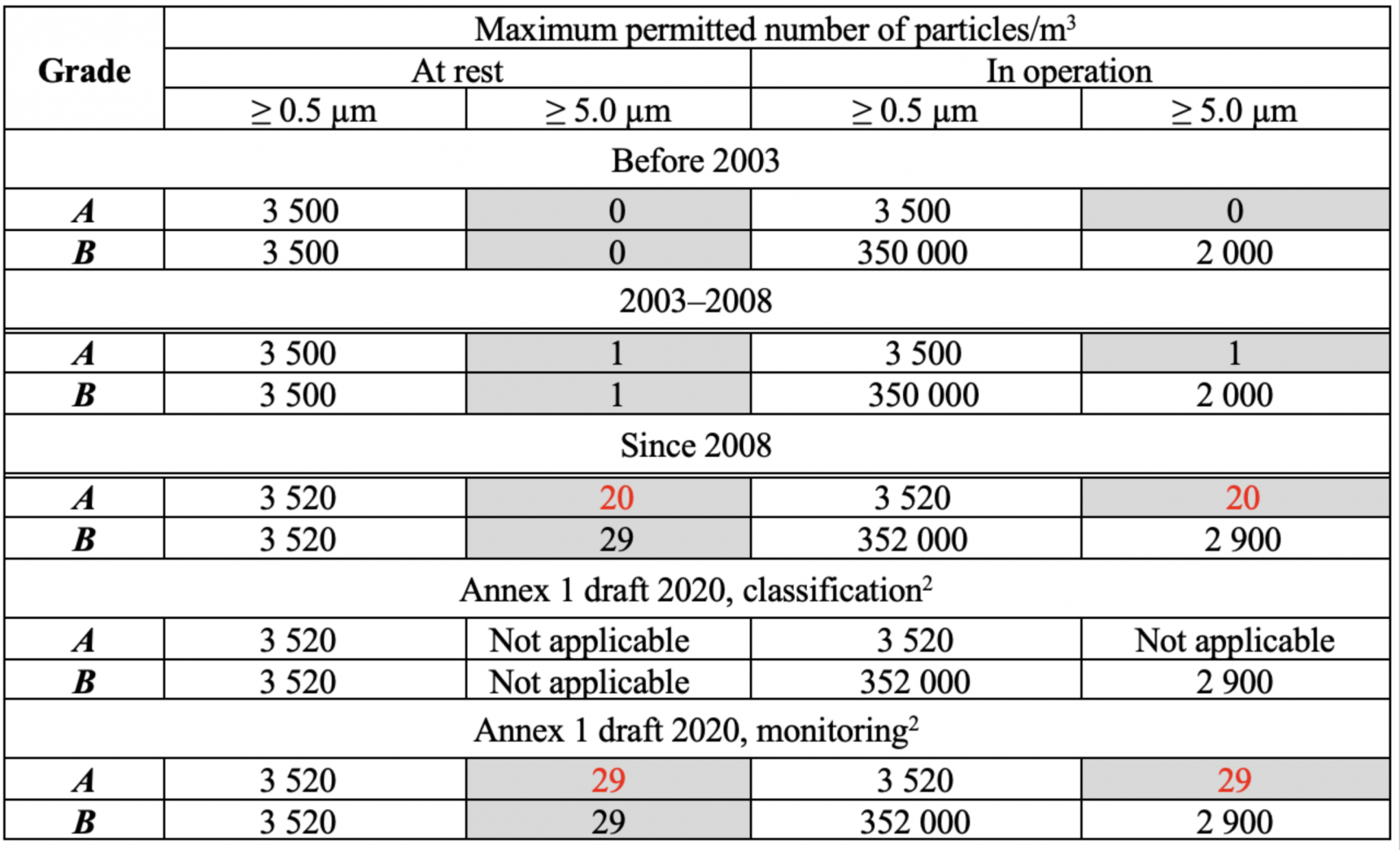
表 1:附件 1 空氣中顆粒物濃度限值的變化,A/B 級。
* Annex 1 draft 2020中分類表的形式與2008年版有所不同,原因不明;本文使用2008表格
2008 年,對於靜止和運行中的 A 級,> 5.0 µm 的顆粒限制從 1 變為 20,結果令人困惑:
a) > 5.0 µm 的顆粒限制變得更弱兩級,被宣佈為最重要的 GMP 目的的對質量的關注又如何呢?舊的 0 或 1 限制在很久以前就在良好的潔淨室中實現了,似乎沒有理由使規範變得更糟:
b) ISO 4.8 級出現在 2008 年,由於粒子計數器精度(通常為±20%)和粒子濃度的實際波動,無法區分 ISO 4.8 和 ISO 5 級。
在此之後,歐盟 GMP附件 1 草案變得更糟,將 > 5.0 µm 的顆粒限制增加到 29 個!
符合 ISO 14644-1 標準:
附件 1 在 2008 年和 2020 年草案發生變化的原因似乎是與 ISO 14644-1:1999 標準(2015 年修訂)保持一致,將表 1 中 2003 年至 2008 年期間的顆粒物濃度轉換為 ISO 等級可得出一幅有趣的圖景(表 2)。
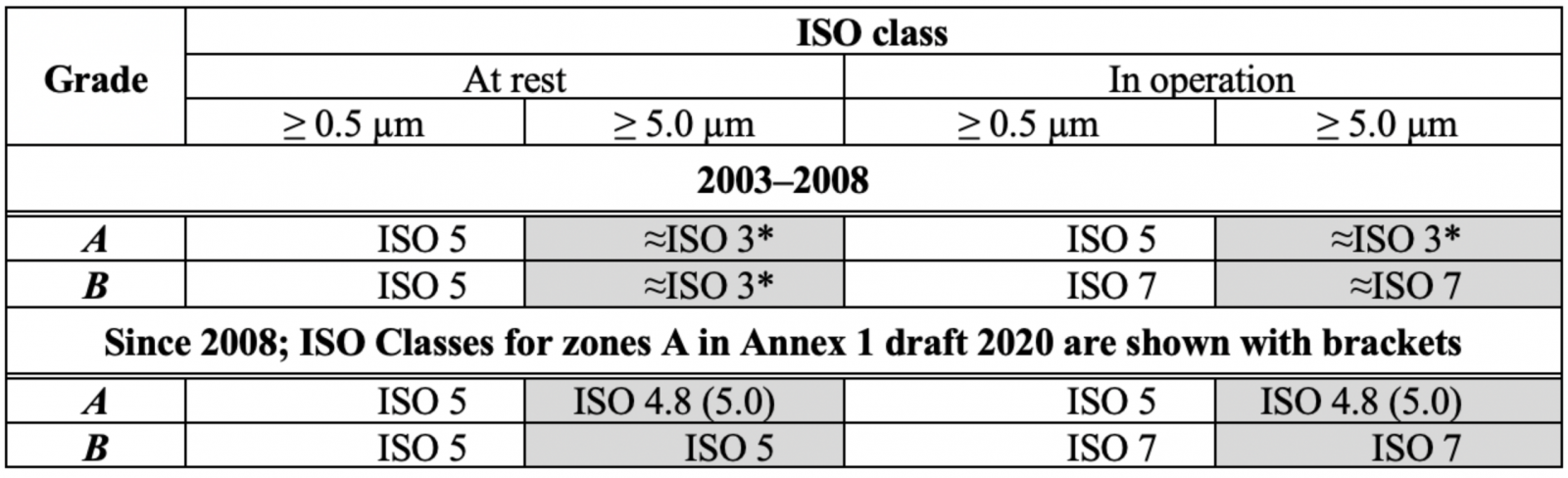
表 2:A/B 級顆粒濃度的附件 1 限值以 ISO 等級表示。* ISO 14664-1 中限制的近似外推
在 2008 年之前,相同佔用狀態(靜止或運行中)的顆粒 > 0.5 µm 和 > 5.0 µm 的 ISO 等級有很大不同,
顆粒 > 0.5 µm 的 A/B 級有 ISO 5 級,ISO 3 級(如果要對 > 5.0 µm 的顆粒進行一些推斷)。
這似乎效果不佳,因此出現了協調的想法,根據 ISO 14644-1:1999,ISO 5 級被指定為靜態 B 級,限制為 29 個 > 5.0 µm/m3 的顆粒,A 級 20 個顆粒 > 5.0 µm/m3 的限制被專家武斷地採用,因為它“應該比 B 級更嚴格”,於是 ISO Class 4.8 出現了。
但潛在的問題是,沒有人問簡單的問題:
ISO 14644-1 表是否以某種方式反映了現實,或者這只是一個抽象的介紹?
它可以用來為同一設施設置不同粒徑的限制嗎?附件 1 中 B 級“靜止”粒子濃度之間的比率為:
Cn>0.5 µm/Cn>5.0 µm = 3500/29 ≈ 121
對潔淨室的測試給出了該比率的實驗數據,對於“靜止”和“運行”佔用狀態,該比率大約在 5:1 和 1000:1 之間,在大多數情況下,B 區的比率 Cn>0.5 µm/Cn>5.0 µm ≈ 5-10。
英國著名 GMP 專家 John Sharp 也提出了類似的結果,當然,從來沒有打算設置不同粒徑的粒子濃度之間的相關性,但是 ISO 表看起來令人印象深刻,但沒有人很好地解釋它的來源,這造成了誤解。
ISO 14644-1分類表的由來
那麼ISO分類表是怎麼出現的呢?著名的美國聯邦標準209在1960年代初期就給出了潔淨室的分類,等級編號表示每 1 立方英尺空氣中尺寸 > 0.5 µm 的顆粒的最大允許濃度(等級 1;10;…100 000),它很簡單,可能是有史以來最好的潔淨室分類系統,它基於對數線性原理,該原理可以表示為閾值 0.5 µm 的圖形上的一條線,
ISO 14664-1 基於日本標準 JIS B 9920:1989,並使用公式 (1) 表示的相同對數線性近似,這個公式是人為的,這可以追溯到美聯標準。
209 對於 > 0.5 µm 的顆粒,將立方英尺轉換為立方米並使用數字序列 1;10…. ,因此單個粒徑 > 0 的對數線性模型,5 µm 散佈到不同的粒徑,每個考慮的顆粒尺寸 D 的最大允許顆粒濃度 Cn 由以下等式確定:
CN = 10否x (1)
在哪裡
N…. 是 ISO 等級編號,
D…是所考慮的粒徑,單位為 µm。
0.1…是一個常數,量綱為 µm。
該公式從不代表實際條件下的粒子濃度。
ISO 14644-1 分類表僅適用於給定應用的一種粒徑(閾值),最後,數字如何出現並不那麼重要。
一個共同的協議是要點,但是圖片正在更改以指定同一潔淨室中不同粒徑的限制,結果可能是不同的清潔等級,這些差異可能是數量級的,這絕對是胡說八道。
當然可以指定一個無意義的,但是,您如何實際實現這種無意義呢?你如何設計和建造一個潔淨室,比如說 ISO 6 級用於顆粒 > 0.5 µm 和 ISO 3 級用於顆粒 > 0.2 µm?沒人知道,如果您嘗試為同一個潔淨室指定兩個不同的等級,那麼在一種情況下可能會有很大的差距,而在另一種情況下可能會有非常嚴格的條件。
您如何解釋不同粒徑的清潔度等級標準?又沒有回答。
2020 年附件 1 草案第 4.29 項建議測量 > 1.0 µm 的顆粒,但不設置限制並解釋其用途,混亂是勝利。
MPPS點
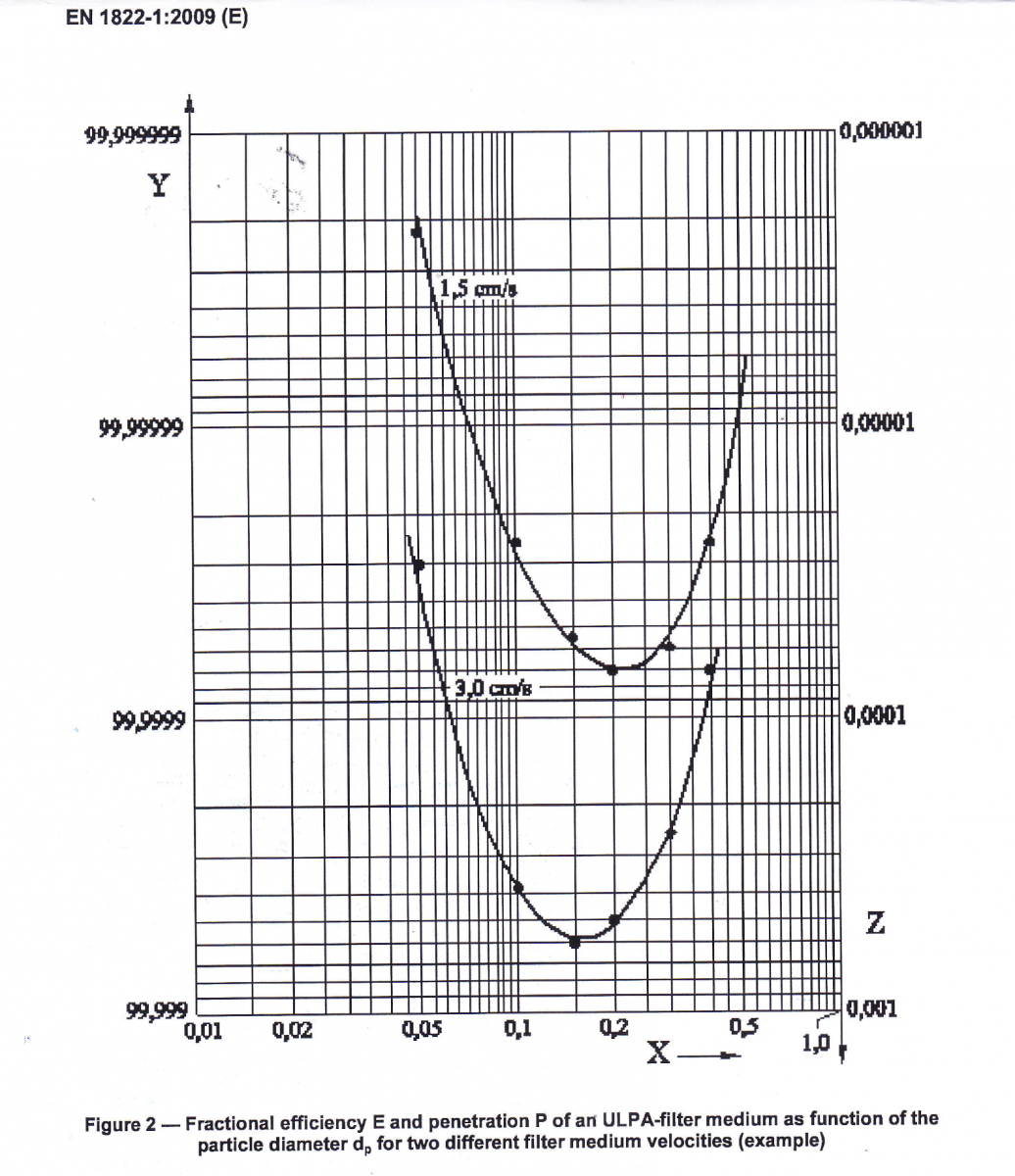
圖 1:對於兩種不同的過濾介質速度,ULPA 過濾介質的分數效率 E 和穿透 P 作為粒徑 dp 的函數(示例)
為不同的顆粒大小設置顆粒濃度限值也取決於空氣過濾器的效率,這種效率極大地取決於顆粒大小,過濾效率曲線有一個最小值,稱為最易穿透粒徑 (MPPS)。
此功能基於基本過濾效果,是自然法則。
通常 HEPA/ULPA 過濾器的 MPPS 點在 0.2 和 0.5 µm 之間,圖 1(圖 2 來自 EN 1822-1:2009),對於較大的顆粒,過濾效率會迅速提高,對於 >5.0 µm 的顆粒,它超過許多數量級。
因此,對 0.5 µm 的顆粒進行測試可以確保可以攜帶微生物的 > 5.0 µm 顆粒的限值將得到滿足,並且具有很大的冗餘性。
測試與監控
附件 1 草案對路由監控設置了比啟動測試更嚴格的要求,這是對常識和慣例的暴力,初始測試應始終比監控更詳細。
結論:
1. 對於 GMP 目的,僅控制尺寸 > 0.5 µm 的顆粒就足夠了。
2. 通常,為同一設施指定不同粒徑的清潔度等級是沒有意義的。一個尺寸就夠了。
3. 已經有人在沒有任何邏輯的情況下推動 GMP 變得更加複雜。
儀器耗材類相關小知識
腳踏黏墊的作用?
