
一、 前言:無菌製程的「隱形防線」
在製藥領域,任何細微的污染都可能導致災難性的後果。根據 FDA 與歐盟 GMP 的規範,無菌藥品的生產必須在極度受控的環境中進行。其中,非活性微粒(Non-viable particles)的監測是評估環境潔淨度、驗證空調系統(HVAC)效能以及監控生產過程污染風險的核心指標。
隨著 2022 年新版 EU GMP Annex 1 正式生效,全球製藥業對於無菌區(特別是 Grade A 與 Grade B)的監測要求已從「抽樣檢查」進化為「基於風險的持續監控」。微粒計數器不再只是檢測工具,而是無菌保證體系中的「即時哨兵」。
二、 法規核心:FDA 與 GMP 對無菌區(A/B 級)的嚴格要求
在製藥廠的潔淨分級中,Grade A(與產品直接接觸的關鍵區域)與 Grade B(後台支撐區域)的規範最為嚴苛。
1. 連續監控(Continuous Monitoring)的要求
新版規範強調,在關鍵的 Grade A 區域,必須在整個生產過程中(包括設備組裝)進行連續的空氣微粒監測。這意味著儀器必須具備高穩定性,並能與廠務監測系統(FMS)整合,確保數據不間斷。
2. 粒徑規格的選取
法規明確要求監測 0.5 µm 與 5.0 µm 的粒子濃度。雖然新版 EU GMP Annex 1 移除了 5.0 µm 作為 Grade A 區域分級標準的要求,但在「風險評估」與「動態監控」中,5.0 µm 粒子的異常增加仍被視為設備磨損或人員介入的重要預警訊號。
3. 數據完整性(ALCOA+)
FDA 對於電子紀錄的要求極高。微粒計數器產出的數據必須符合可追溯、即時、原始且準確的原則。這對現場使用的計數器提出了技術挑戰:它是否具備審計追蹤(Audit Trail)功能?是否能防止數據被篡改?
三、 從靜態到動態:動態(Operational)監測的應用與挑戰
GMP 規範將監測狀態分為「靜態(At-rest)」與「動態(Operational)」。而對於製藥廠而言,真正的考驗在於動態監測。
1. 動態監測的真諦:監控人的變因
無菌區最大的污染源通常是「操作人員」。在動態生產過程中,微粒計數器必須能即時捕捉到因人員動作、物料傳送或設備運作所產生的粒子波動。如果計數器感度不足或取樣頻率不夠,就可能錯失潛在的污染事件。
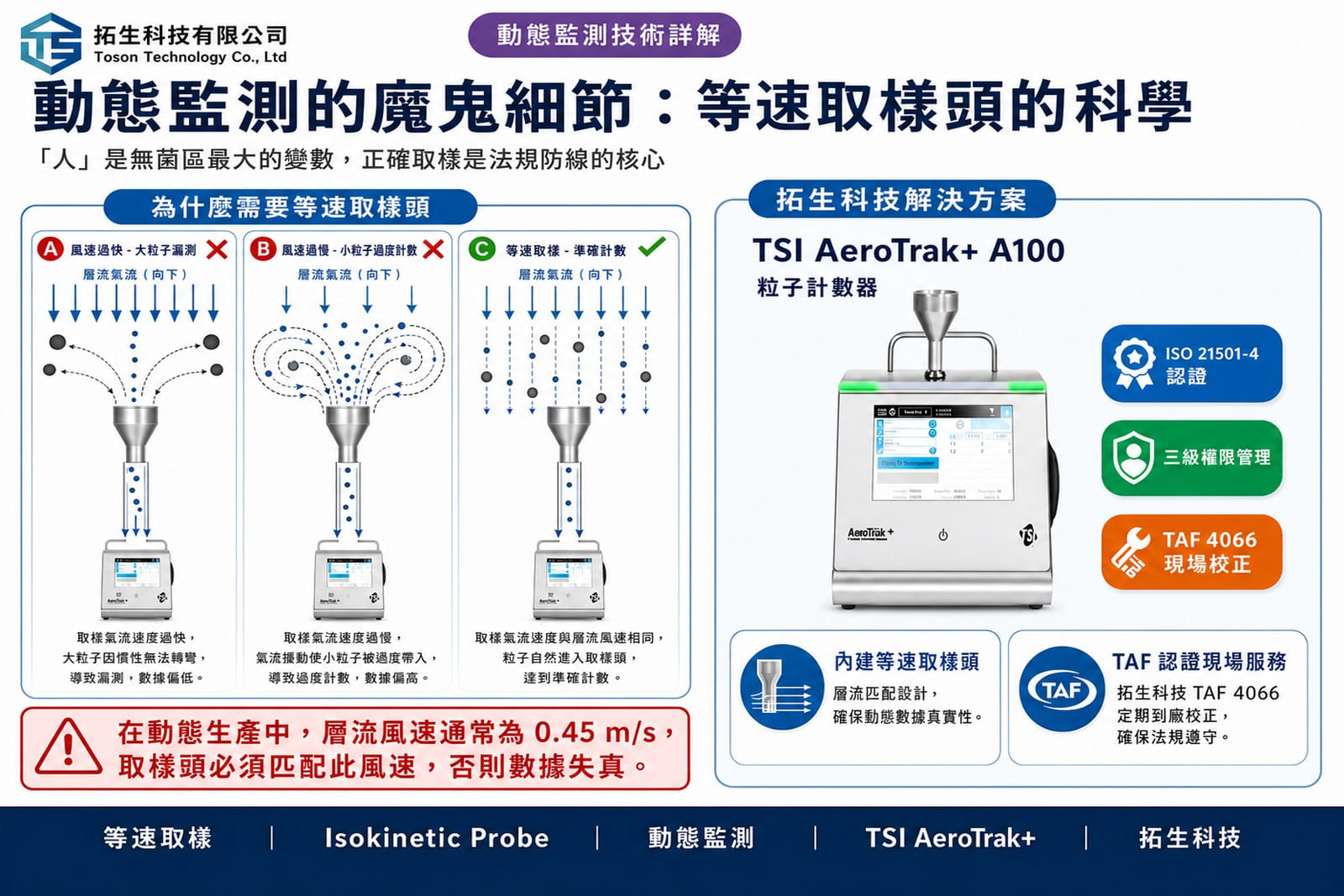
2. 取樣位置與等速取樣(Isokinetic Sampling)
在動態環境下,氣流會受到設備幾何形狀與操作動作的干擾。FDA 要求取樣頭必須位於「風險最高的點」,且必須使用等速取樣頭(Isokinetic Probe),以確保取樣口風速與潔淨室層流風速一致,避免因慣性效應導致大粒徑粒子(如 5.0 µm)的漏測或小粒徑粒子的過度計數。
3. 異常觸發與警報策略
動態監測的目的在於「即時反應」。製藥廠必須根據驗證數據設定「警示限度(Alert Level)」與「行動限度(Action Level)」。一旦數據超標,系統需立即觸發警報,引導操作員執行預定的偏差處理(Deviation Handling),而非等到批次生產結束後才發現問題。

四、 拓生科技:助力製藥廠對標國際標準
身為環境監測的專業夥伴,拓生科技針對製藥 GMP 的嚴格要求,提供全方位的技術支援:
- 符合 Annex 1 要求的高精度儀器: 我們代理的 TSI AeroTrak™+ A100 系列微粒計數器具備 1.0 CFM (28.3 LPM) 或 100 LPM 的穩定流量,並完全符合 ISO 21501-4 校正規範,確保數據具備科學公信力。
- 數據完整性與 21 CFR Part 11 支援: 儀器內建三級權限管理與 Audit Trail 功能,並協助客戶將現場計數器整合至中央監測系統,確保符合 FDA 21 CFR Part 11 要求。
- TAF 認證的現場驗證服務: 我們的技術團隊可提供符合規範的現場校正與取樣點評估,協助藥廠建立科學的風險監測計畫。
五、 結語:合規,是為了更安全的藥品
在 FDA 與 GMP 的嚴格監管下,微粒監測已不再是簡單的數字遊戲,而是製藥企業技術實力的體現。透過導入先進的動態監測技術與嚴謹的校正體系,藥廠不僅能順利通過國內外查核,更能從源頭守護病患的用藥安全。
延伸閱讀
【生物醫藥遵循 PIC/S GMP 或 EU GMP】設備採購指南:法規導向的微粒監控決策